This page contains a list of programs/servers developed in Xu group. Please feel free to download/use them. However, they are provided as is and there is no warranty.
1) PhyCMAP: a protein contact
map prediction server makes use of both evolutionary and non-evolutionary information. See our ISMB2013
presentation for a brief introduction and our paper http://arxiv.org/abs/1308.1975 for
more technical details and results. PhyCMAP first uses statistical machine learning to
predict contact probability and then integer programming to select the most probable contacts subject to physical constraints.
The evolutionary information used by PhyCMAP includes sequence profile and residue co-evolution. In particular, PhyCMAP uses the powers of the mutual information
matrix (i.e., MI, MI2, MI3,..., MI11) to account for the chaining effect of residue couplings and contrastive MI to
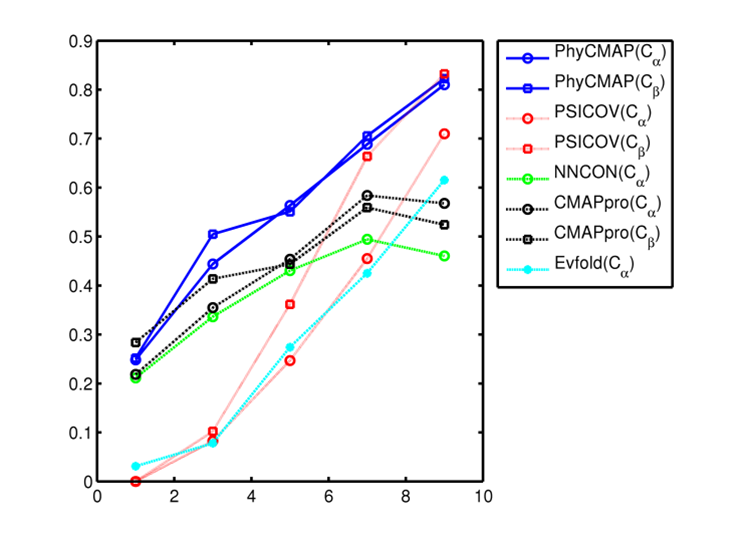
remove local bias. The below figure shows that PhyCMAP outperforms existing methods regardless of the number of
non-redundant sequence homologs available for a protein under prediction.
|
|
This figure shows the relationship between prediction accuracy and the number of non-redundant sequence homologs (Meff). X-axis is log(Meff) and Y-axis is the mean accuracy of top L/10 predicted contacts in the corresponding CASP10 target group. Only medium- and long-range contacts are considered. All the CASP10 targets are divided into groups by log(Meff). Meanwhile PSICOV and Evfold are co-evolution-based method, and NNcon and CMAPpro are supervised machine learning methods. |
2) EPAD: a context-specific distance-dependent
statistical potential for protein study. See our paper http://www.cell.com/structure/abstract/S0969-2126(12)00145-1
for the technical details. The whole package is available at EPAD.tar.gz,
which also includes some APIs (Application Programming Interface) so that you
can easily integrate EPAD into your own projects. A simple user guide is
available at EPAD_guide.html
and the documentation for the EPAD APIs is available at EPAD_API_ref.pdf.
3) DeepAlign: a program for pairwise protein structure alignment.
Different from many other tools, DeepAlign aligns two
protein structures using evolutionary information and beta strand orientation
in addition to geometric similarity. Therefore, DeepAlign
favors the alignment of evolutionarily-related residues and also aligns beta
sheets more correctly than the others. The DeepAlign
alignments are also much more consistent with manual alignments than the
others. See our paper http://www.nature.com/srep/2013/130314/srep01448/full/srep01448.html
for technical details. The whole package is available at DeepAlign_V1.13.
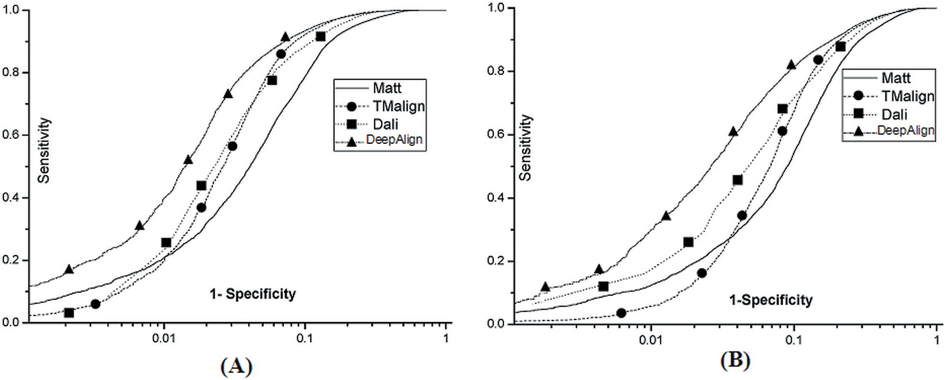
Below see the ROC curves of DeepAlign, DALI, Matt and
TMalign on (A) SABmark-sup
and (B) SABmakr-twi.
4) 3DCOMB: a program for multiple protein structure
alignment. Please download 3DCOMB_exe_V1.06.7z
or 3DCOMB_exe_V1.06.rar.
5) RaptorX: a server and a standalone program for protein
sequence-structure alignment and structure prediction by threading, available
at http://raptorx.uchicago.edu. RaptorX excels at the alignment of hard targets, which have
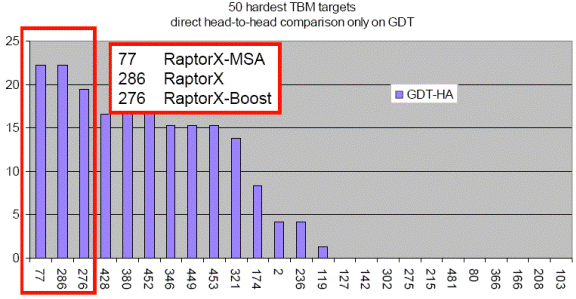
less than 30% sequence identity with solved structures in PDB. As shown in the
below figure, blindly tested on the 50 hardest CASP9 template-based modeling
targets, RaptorX outperforms all the CASP9 participating
servers including those using consensus and refinement methods.
|
|
This figure shows the performance of top 25 groups on the 50 hardest CASP9 TBM targets. X: group numbers. Y: the number of top 25 groups outperformed by a given group. The figure is taken from the CASP9 assessor’s presentation at http://predictioncenter.org/casp9/doc/presentations/CASP9_TBM.pdf. |
6) RaptorX-SS8: a program for protein 3-class and 8-class
secondary structure prediction. Please download the code raptorx-ss8.tar.gz
and readme
file. RaptorX-SS8 is also integrated into the
RaptorX server.
7) RaptorX-FM: a program for fragment-free protein folding.
Please download code RaptorXFM.tar.gz
and readme
file.
8) CNF: a program for conditional neural fields, a
variant of conditional random fields.
9) TreePack: a program for protein side-chain packing. It is
available at TreePack.